
CAZy数据库简介
CAZy 全称为Carbohydrate-Active enZYmes Database,碳水化合物酶相关的专业数据库,内容包括能催化碳水化合物降解、修饰、以及生物合成的相关酶系家族。其包含五个主要分类:糖苷水解酶(Glycoside Hydrolases, GHs)、糖基转移酶(GlycosylTransferases, GTs)、多糖裂解酶(Polysaccharide Lyases, PLs)、糖酯酶(Carbohydrate Esterases, CEs)和氧化还原酶(Auxiliary Activities, AAs)。此外,还包含与碳水化合物结合结构域(Carbohydrate-Binding Modules, CBMs)。五大分类和一个结构域下,都分别建立了多个Family。
CAZy数据库的准备
在进行预测之前需要准备数据库,CAZy貌似没有提供FASTA格式的序列数据库,而仅提供了序列的Assenssion number,需要我们自己从NCBI数据库中下载序列。下载方法参照我之前的文章《根据assession number批量从NCB下载数据》,在文章中提供了下载CAZy序列的方法和脚本,此处不再赘述。
在上一篇文章结尾获得的“All.sequences.fas”文件包含了所有的CAZy数据库序列,在正式预测之前需要完成数据库的格式化。后面我们将通过Diamond软件从基因组中预测CAZy蛋白,因此采用Diamond格式化数据库。
序列预处理
不知道什么原因,下载的序列存在两个问题,其一,下一条序列的ID连接着上一条序列的末尾,没有断行;其二,序列中存在着一段网页代码。因此,需要分两步进行修正。
解决断行问题
撰写脚本“add_linebreak.pl”,内容如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16#!/usr/bin/perl
use strict;
use warnings;
# Author: Liu hualin
# Date: Sep 30, 2021
local $/=">";
open IN, "All.sequences.fas" || die;
open OUT, ">CAZy.fas" || die;
<IN>;
while (<IN>) {
chomp;
print OUT ">$_\n";
}
close IN;
close OUT;将脚本与”All.sequences.fas”放在同一目录下,在终端或者命令行中运行如下命令,得到“CAZy.fas”。
1
perl add_linebreak.pl
删除无关内容
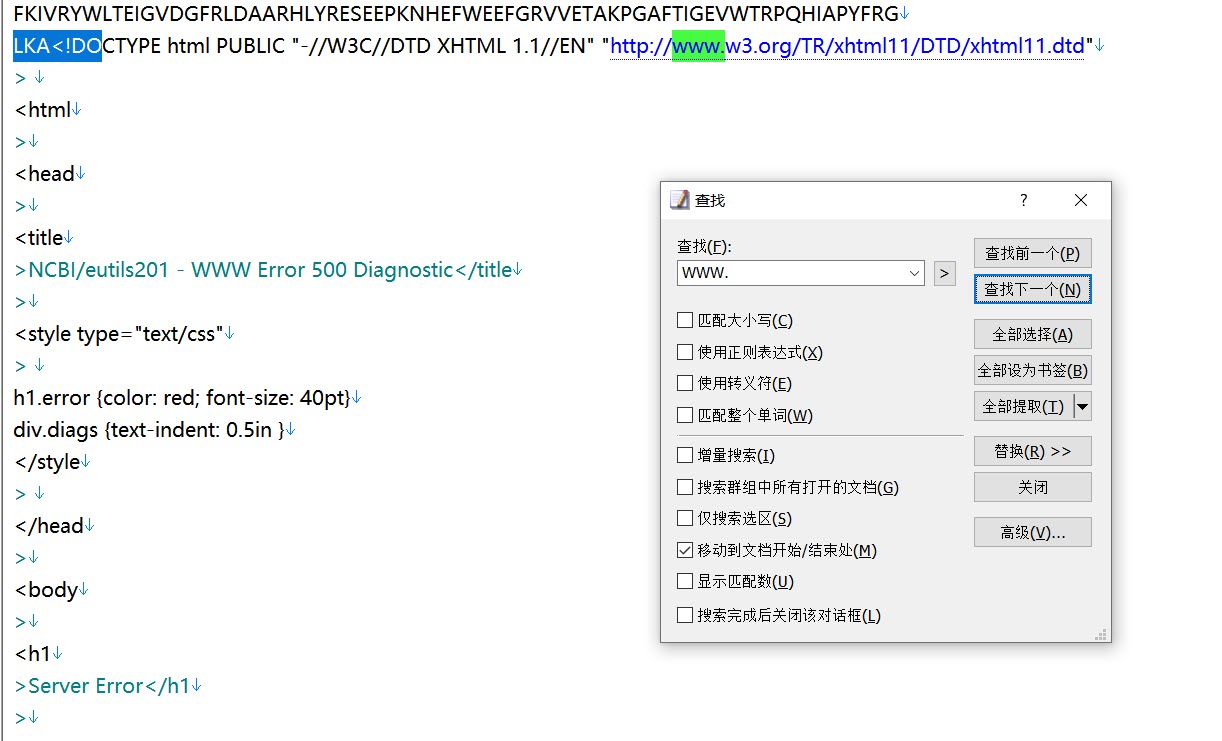
用EmEditor软件打开CAZy.fas,Ctrl+F调出查找功能,搜索“www.” 可以看到如下内容,手动将其删除,并保存文件。
构建Diamond数据库
1 | diamond makedb --in CAZy.fas -d CAZy |
开始序列比对
当然,我们选择用Perl进行批量比对
1 | #!/usr/bin/perl |
将上述代码复制到文件中,命名为“run_diamond_CAZy.pl”,将其和序列文件放在同一目录下,并在终端中输入如下命令,完成分析,得到“*.CAZy.diamond”:
1 | perl run_diamond_CAZy.pl |
比对结果过滤
在比对过程中我们控制了evalue和query coverage,但是没有控制identity。但是很多时候,需要设定一个identity的阈值,低于阈值的比对将会被删除,该步骤可以将比对结果拷贝到Excel中根据identity排序,手动删除阈值以下的行,然而我选择用Perl批处理。
1 | #!/usr/bin/perl |
将上述代码复制到文件中,命名为“filter_cazy_diamond.pl”,将其和上一步产生的文件放在同一目录下,并在终端中输入如下命令,完成过滤,保留identity >= 40% 的行,得到“*.CAZy.diamond.filtered”。
1 | perl filter_cazy_diamond.pl |
你以为完了?还得mapping!
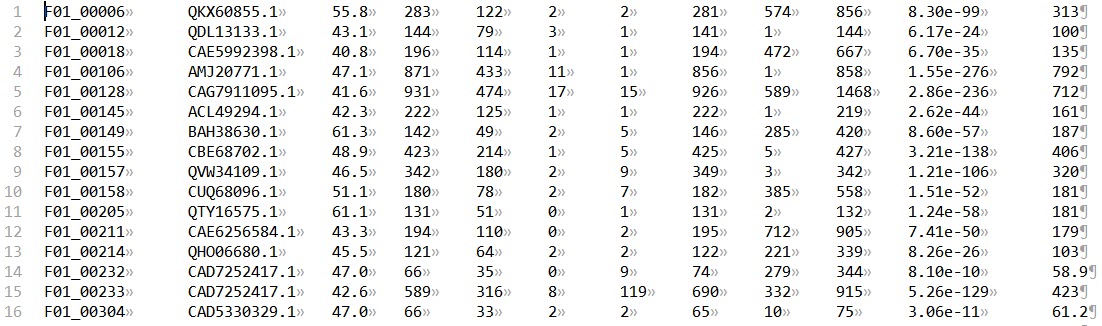
得到的结果如下图所示,第二列的Hits是NCBI的Assession number,我们根本只知道这是什么CAZy家族,因此需要mapping!
回头找到我们下载的cazy_data.txt,里面保存的是CAZy家族与Assession number的对应关系。比较闲的兄弟可以用查找-复制-粘贴的方法将“*.CAZy.diamond.filtered”中的Assession number替换为CAZy家族。我为比较忙的兄弟准备了下面的代码,批处理。不过我输出的是一个矩阵。
1 | #!/usr/bin/perl |
将上述代码复制到文件中,命名为“assession2cazy.pl“,将其和”cazy_data.txt“,及上一步产生的文件“*.CAZy.diamond.filtered”放在同一目录下,并在终端中输入如下命令:
1 | perl assession2cazy.pl |
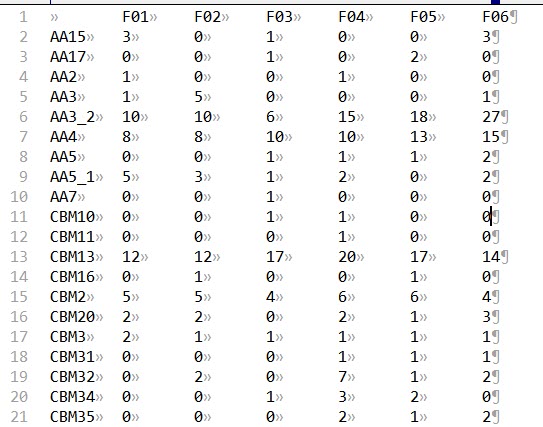
得到一个矩阵“CAZy.Matrix.txt”,内容如下,行为CAZy家族,列为基因组/样本名。拿到本文件后,可以做热图看CAZy家族在各样本中的分布情况,然而这个热图将会比鞋帮子脸还要长,可读性不高,因此我选择将这些family合并为大类,生成一个新的矩阵。
二话不说,上代码。
1 | #!/usr/bin/perl |
将上述代码复制到文件中,命名为“cazyfamily2categories.pl”,将其和上一步产生的文件“CAZy.Matrix.txt”放在同一目录下,并在终端中输入如下命令,得到文件“CAZy.Category.Matrix.txt”。
1 | perl cazyfamily2categories.pl |
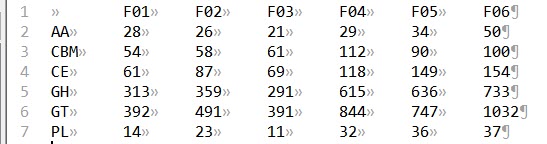
接下来是要做柱状图还是heatmap,就随便了。
脚本获取
关注公众号“生信之巅”,聊天窗口回复“9052”获取下载链接。
 |
 |
敬告:使用文中脚本请引用本文网址,请尊重本人的劳动成果,谢谢!



